Glucose is central to all metabolism. Every human cell type uses glucose as a source of energy and as a source of carbon skeletons for the synthesis of other compounds. Normally, all cells need a continuous supply of glucose. To meet this need, blood glucose concentration is maintained within a relatively narrow range of approximately 3.6 - 5.8 mM (64.8 - 104.4 mg/dL), independent of dietary supply, i.e., in both the well-fed state and the fasted state. This process is known as glucose homeostasis. Hypoglycemia (low blood glucose) is prevented by the release of glucose from stores of glycogen in the liver (glycogenolysis), by the synthesis of glucose in the liver and to a lesser extent in the kidney from non-carbohydrate carbon sources (gluconeogenesis), in particular from lactate, amino acids, and glycerol, and by the release of fatty acids from adipose tissue (lipolysis) to provide an alternate source of energy. Hyperglycemia (high blood glucose) is prevented by glycogenesis (glycogen synthesis from glucose), mainly in the liver and muscles, and by the conversion, in the liver, of glucose to triacylglycerols (fats), which are transported to and stored in adipose tissue.
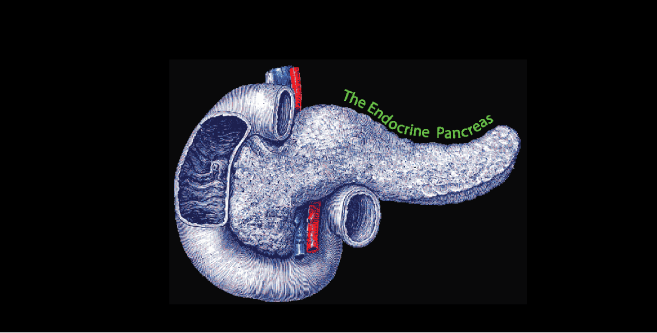
The Pancreas is a Glucose Sensor that secrets either Insulin or Glucagon depending on the blood [glucose] that it senses.
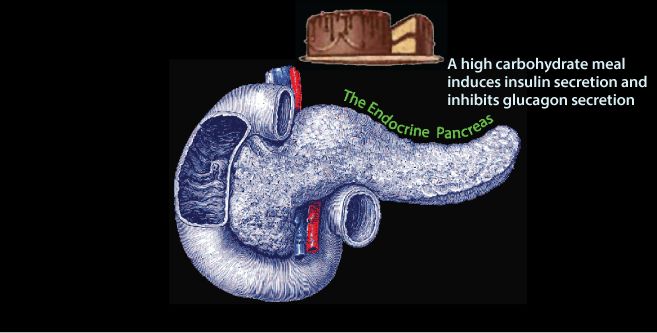
Glucose enters the blood from the intestine after a meal. The increased blood [glucose] is sensed by the pancreas, which secretes insuline in response.
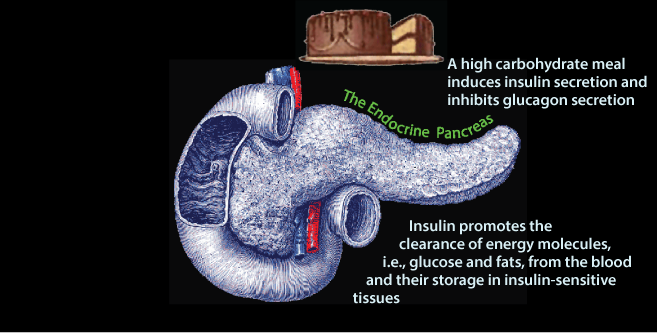
In response to insulin signaling, excess glucose is removed from the blood. The liver and skeletal muscle, and to a lesser extent adipose tissue are the predominant tissues that clear glucose from the blood. The liver and skeletal muscle store glucose as glycogen. The liver uses glucose carbon to systhesize triacylglycerol (fat), which it exports in very low density lipoproteins (VLDL) for storage in adipose tissue. Adipose tissue uses glucose to derive glycerol to store the fatty acids delivered to it by VLD in adipose triacylglycerol.
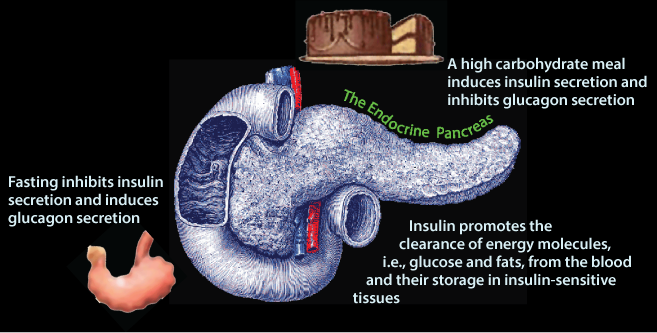
After the last food has been absorbed from the intestine (note the empty stomach indicated by its “puckering” !) and excess glucose has been cleared from the blood, normal blood [glucose] is maintained from stored glycogen and gluconeogenesis. In response to decreased blood [glucose] pancreatic β cells decrease insulin secretion and the pancreatic α cells increase glucagon secretion.
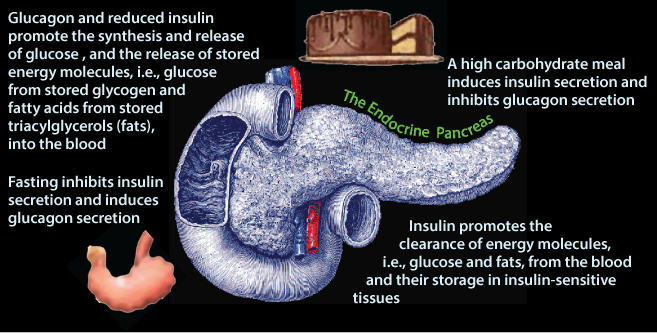
Glucagon signaling and the absence of insulin signaling cause the liver to release glucose from stored glycogen and de novo glucose synthesis 9gluconeogenesis) into the blood, and adipose tissue to release fatty acids from the hydrolysis of stored triacylglycerol into the blood.
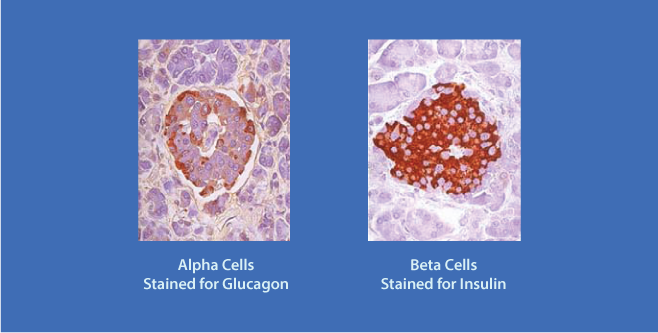
The Pancreatic Islets secrete Insulin and Glucagon. The islet α cells ( rust color ), located at the periphery of the islet secrete Glucagon; the β cells ( rust color ), located more centrally, secrete Insulin.
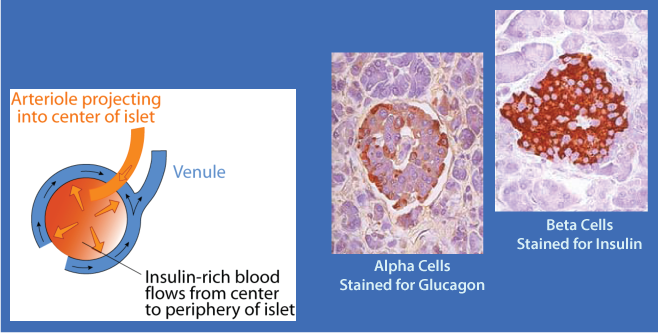
Pancreatic α cells have insulin receptors. β cell-secreted insulin acts on the α cell insulin receptors to inhibit glucagon secretion. The flow of blood in the islet is from the center outward. The glucagon-secreting α cells are located at the periphery of the islet and receive insulin-rich blood when insulin is secreted, thereby preventing the simultaneous secretion of both insulin and glucagon when blood [glucose] is increased.
Insulin and glucagon are the two major hormones that regulate fuel metabolism and storage to ensure that cells have a constant supply of glucose, fatty acids, and amino acids for ATP generation and cellular maintenance. The balance between insulin and glucagon, ensures blood glucose homeostasis by regulating carbohydrate, lipid and amino acid metabolism according to the needs and capacities of individual tissues. The release of insulin from the β cells of the pancreas is regulated primarily by the concentration of blood glucose.
The fed state; insulin predominates:
Major sites of insulin action on fuel metabolism
+, stimulated by insulin; -, inhibited by insulin.
Insulin concentration in the blood reaches a maximum approximately 30-45 minutes after a high carbohydrate meal. Insulin, released in response to carbohydrate ingestion, promotes glucose utilization as fuel and glucose storage as fat and glycogen. Insulin is also the major anabolic hormone of the body; it increases amino acid uptake into tissues, protein synthesis, and cell growth, as well as fuel storage. The blood concentration of insulin declines approximately 120 minutes after a meal as blood glucose concentration declines due to glucose uptake and use by tissues. The blood concentration of glucagon, the major counter-insulin regulatory hormone, is decreased in response to a carbohydrate meal and increases during fasting. The lowest concentration of glucagon in the blood occurs after a high carbohydrate meal, and increases as blood glucose concentration decreases.
Glucagon functions to maintain fuel availability in the absence of dietary glucose by promoting glycogenolysis and gluconeogenesis. The release of glucagon from the α cells of the pancreas is controlled primarily through suppression by glucose and insulin.
The fasted/starved state; glucagon predominates:
Major sites of glucagon action on fuel metabolism.
+, stimulated by glucagon; -, inhibited by glucagon. NOTE that in adipocytes low insulin, not increased glucagon, is the regulator of fatty acid generation from triacylglycerols for export to the blood. Also, muscle does not respond to glucagon - it has no glucagon receptors.
Decreased blood insulin, at times when blood glucagon is increased, stimulates the mobilization of fatty acids from adipose tissue stores of triacylglycerols. Because the effects of glucagon are opposed by insulin, the simultaneous secretion of insulin and suppression of glucagon secretion by a high carbohydrate meal provides integrated control of the metabolism of carbohydrate, fat and protein.
Epinephrine, the acute stress, or “fight or flight” hormone and cortisol, a glucocorticoid (steroid hormone), are synthesized in the adrenal gland and released, respectively, in response to acute or chronic stress. They are counter-insulin regulatory hormones that have effects on fuel metabolism opposite to those of insulin. They function to increase fuel availability so the body can respond to a variety of different stressful conditions.
Fuels taken in the diet in excess of immediate needs are stored and the appropriate fuel is mobilized when demand occurs. These processes maintain metabolic homeostasis so that tissues have a constant source of fuels from which to synthesize ATP for the maintenance of normal cell function and growth. For example, if dietary glucose is not available in sufficient quantities that all tissues can use, fatty acids are mobilized and can be used by muscle and other tissues, and the liver can convert fatty acids to ketone bodies, which the brain and other tissues can use for their energy needs. The brain can use ketone bodies for approximately 80% of its energy needs, but cannot use fatty acids, and must always have glucose for approximately 20% of its energy needs. The use of fatty acids for energy spares glucose for use by the brain and other glucose-dependent tissues (such as red blood cells).
Insulin and glucagon are synthesized in the endocrine pancreas and secreted into the hepatic portal vein. Active insulin is composed of two polypeptide chains; the A-chain and the B-chain are covalently linked by two inter-chain disulfide bonds. The A-chain has one additional intrachain disulfide bond. Insulin is synthesized as a preprohormone that is converted to proinsulin in the rough endoplasmic reticulum (RER) and subsequently undergoes further processing steps to yield the mature insulin molecule in the Golgi apparatus.
Insulin is synthesized as a preprohormone that is converted to proinsulin in the rough endoplasmic reticulum (RER). The “pre” sequence at the N–terminal end is cleaved as it enters the lumen of the RER.
Proinsulin folds into its proper conformation and disulfide bonds are formed between specific cysteine residues. It is transported in membrane-bound vesicles to the Golgi complex,
from which it leaves in storage vesicles where it is cleaved by a protease to release the C-peptide fragment and a few small remnants, which have no hormonal activity. Cleavage of the C-peptide decreases the solubility of the insulin, which co-precipitates with zinc ions that are also present in the storage vesicles.
Mechanism of insulin secretion from pancreatic β cells:
Glucose enters the β cells via a glucose transporter, GLUT2, and is phosphorylated by glucokinase to glucose-6-phosphate, which is metabolized by the glycolytic pathway, the TCA cycle, and oxidative phosphorylation to yield ATP
As the β cell [ATP]/[ADP] ratio increases, the activity of a plasma membrane-bound ATP-regulated K+ channel is inhibited,
and the channel closes,
causing membrane depolarization,
which activates a voltage-gated Ca 2+ channel to allow Ca 2+ entry into
the β cell.
The increased intracellular [Ca 2+ ] stimulates the fusion of the insulin
storage vesicles with the plasma membrane, thereby releasing insulin.
and the C-peptide by exocytosis.
Drugs that target insulin secretion to treat type 2 diabetes :
Sulfanylureas and Meglinitides (red asterisk) target separate sites on the potassium channel, causing it to close. Synthetic oligopeptides that mimic Glucagon-Like Peptide-1 (GLP-1) bind to its receptor (green asterisk) to activate signaling events downstream of the receptor that increase insulin secretion through the activity of protein kinase A (PKA).
Factors other than blood [glucose] also modulate insulin release. The pancreatic islets are innervated by the autonomic nervous system, including a branch of the vagus nerve. Neural signals help to coordinate insulin release with the secretory signals initiated by the ingestion of fuels, but signals from the central nervous system are not required for insulin secretion in response to fuel ingestion.
| Regulators of Insulin Secretion From Pancreatic β Cells | |
| Major Regulators | Effect |
| Glucose | + |
| Minor Regulators | |
| Some Amino Acids | + |
| Neural Input | + |
| Gut Hormones | + |
| Epinephrine | - |
| Effect: +, stimules; -, Inhibits | |
Some amino acids can also stimulate insulin secretion, although the amount of insulin released in response to the ingestion of a high-protein meal is very much lower than that released in response to the ingestion of a high-carbohydrate meal. Gastric inhibitory polypeptide (GIP), also called glucose-dependent insulinotropic peptide and glucagon-like peptide-1 (GLP-1), two gut hormones (incretins) released after the ingestion of food, also aid in the onset of insulin release. Epinephrine, which is secreted in response to fasting, stress, trauma and vigorous exercise, decreases the release of insulin, thereby promoting the mobilization of stored fuels for the generation of the energy required to respond to these physiological conditions, rather than fuel storage, as promoted by increased insulin.
Glucagon, a 29 amino acid polypeptide hormone, is synthesized in the α cells of the pancreatic isles of Langerhans by cleavage from a larger preproglucagon protein. Like insulin, it is synthesized in RER and converted to proglucagon as it enters the lumen of the RER. Proteolytic cleavage generates the mature 29-amino acid glucagon and larger glucagon-containing fragments, glucagon-like polypeptides 1 and 2 (GLP-1, GLP-2).
Secretion of insulin and glucagon into the blood after an overnight fast followed by ingestion of 100 grams of protein: Note that insulin secretion is substantially less than that after a carbohydrate meal but glucagon secretion is substantially greater.
| Regulators of Glucagon Secretion From Pancreatic α Cells | |
| Major Regulators | Effect |
| Glucose | - |
| insulin | - |
| Some Amino Acids | + |
| Minor Regulators | |
| Cortisol | + |
| Neural Input (stress) | + |
| Gut Hormones | + |
| Epinephrine | + |
| Effect: +, stimules; -, Inhibits | |
Glucagon secretion is regulated principally by the blood glucose concentration and insulin. Increasing concentrations of each inhibit glucagon secretion. The direction of blood flow in the islets of the pancreas carries insulin from the β cells at the center of the islets to the peripheral α cells, where it suppresses glucagon secretion (see diagram in the clickable "The Endocrine Pancreas" above). Some hormones stimulate glucagon secretion, particularly, catecholamines (including epinephrine), cortisol, and some gastrointestional hormones. In addition, some amino acids stimulate glucagon secretion. Thus, the increased concentration of glucagon that would be expected in the fasted state does not decrease after a high-protein, low glucose meal, and the increased glucagon stimulates gluconeogenesis (predominantly from the dietary amino acid carbon skeletons) to maintain blood glucose homeostasis. The relative concentrations of insulin and glucagon in the blood after a mixed meal depend on the composition of the meal, because glucose stimulates insulin secretion and some amino acids stimulate glucagon secretion.
To affect the flux of substrates through a metabolic pathway a hormone must be able to change the rate at which that pathway proceeds. It must affect the activity of specific enzymes or transport proteins to either cause the amount of a substrate to increase (if substrate supply is rate-limiting), change the conformation at the active site by phosphorylating the enzyme, change the concentration of an allosteric effector of the enzyme, or change the amount of an enzyme by inducing or repressing its synthesis or by changing its turnover rate or cellular location. The effects mediated by phosphorylation or changes in the kinetic properties of an enzyme occur rapidly, within minutes, but it may take hours for the induction or repression mechanisms that change the amount of an enzyme in a cell.
Hormones initiate their actions on target cells by binding to specific receptors or binding proteins. The action of polypeptide hormones, such as insulin and glucagon, and catecholamines, such as epinephrine and norepinephrine, is mediated through their binding to the extracellular domain of their cognate receptors in the plasma membrane. This binding activates the receptor to transmit a signal through the plasma membrane to an intracellular “second messenger” (the extracellular hormone being the “first messenger”). Thus, the hormone remains extracellular, but transmits its signal to the inside of the cell. In contrast, steroid hormones, such as cortisol, pass directly through the plasma membrane and exert their effects by binding to, and activating, their cognate intracellular receptor proteins.
The mechanism by which a hormone signal ultimately affects the regulation of a cellular pathway(s) is known as signal transduction. The three basic types of signal transduction for hormones that bind to receptors in the plasma membrane are (1) receptor coupling to adenylate cyclase, which produces cyclic AMP (cAMP), (2) receptor kinase activity – the receptor has a protein kinase activity that becomes active when hormone binds, and (3) receptor coupling to the hydrolysis of phosphatidylinositol bisPhosphate (PIP2) from membrane phospholipids.
The insulin receptor is a member of the tyrosine kinase family of receptors. It is composed of four polypeptide subunits, two α−β pairs (α2β2) that span the plasma membrane. The two α subunits bind insulin, and the two β subunits each have a tyrosine kinase domain. Upon insulin binding to the two α subunits, the tyrosine kinase activity in each of the β domains (indicated by a group of three diagonal lines in the diagram) phosphorylates specific tyrosine residues in the opposite β subunit (receptor autophosphorylation, indicated by crossed arrows). This autophosphorylation activates the receptor, which then binds the insulin receptor substrate (IRS) protein and phosphorylates it at multiple sites to create multiple binding sites for different proteins that contain “sarc homology 2” (SH2) domains (concave surfaces on the proteins that bind to the phosphorylated IRS, i.e., Grb2, PLCγ, PI3-kinase). These IRS binding proteins are also associated with phosphatidylinositol phosphates (PIP), constitutents of phospholoipids in the cytosolic face of the plasma membrane. One of the sites binds Grb2, leading to activation of the Ras protein (not shown) and the downstream MAP kinase pathway that participates in several cell functions, including cell proliferation. Grb2 is anchored to phosphoinositol-3-4-5-trisP in the plasma membrane through its pleckstrin homology (PH) domain (not indicated). Phosphoinositide 3-kinase (PI3-kinase - indicated in color) binds at another phosphotyrosine site and is activated. At a third site, phospholipase Cγ (PLCγ) binds and is activated. It cleaves the phosphorylated inositol moiety from the membrane phospholipids to yield diacyl glycerol and inositol tris-phosphate (IP3), which act as second messengers to regulate various intracellular signaling pathways.

The signaling pathway initiated by the insulin receptor complex involving PI3-kinase leads to activation of protein kinase B (PK B), also called Akt, and atypical protein kinase C (aPK C), serine-threonine protein kinases that mediate many of the downstream effects of insulin on glucose metabolism, including GLUT4 mobilization to the plasma membrane, activation of protein phosphatases that reverse the effects of glucagon, e.g., the synthesis of glucokinase, phosphoenolpyruvate carboxykinase, fructose-1,6-bisphosphatase and glucose-6 phosphatase. Phosphorylation by PK B/Akt and/or aPK C also mediates the affects of insulin on several other cellular processes, including protein synthesis, cell growth, cell cycle entry and cell survival.
The insulin receptor is a tetramer of two α subunits and two β subunits. The α subunits are exposed to the extra-cellular space where they can bind insulin. The β subunits pass through the plasma membrane into the inside of the cell. The β subunits have a protein tyrosine kinase (receptor tyrosine kinase).
Insulin binding activates the tyrosine kinase activity of the receptor β subunits, which cross-phorshorylate the opposite β subunit on specific tyrosine residues.
The receptor tyrosine phosphorylation allows the recruitment of the insulin receptor substrate protein - IRS,
which is phosphorylated on specific tyrosine residues by the insulin receptor tyrosine kinase. Note that in type 2 diabetes this phosphorylation of IRS is inhibited, contributing to the insulin resistance. Several serine/threonine protein kinases, which are activated by other signaling pathways in response to various physiological stresses that occur in type 2 diabetes, phosphorylate IRS on serine and threonine residues. These serine/threonine phosphorylations inhibit the IRS-activating tyrosine phosphorylations catalyzed by the insulin receptor protein tyrosine kinase.
Phosphatidyl Inositol 3 kinase - PI3 kinase - is recruited by the binding of its "Src Homology" domain to a specific tyrosine-phosphorylated region of IRS.
The recruited and activated PI3 kinase phosphorylates Phosphoinositol-4,5-bis Phosphate (PIP2), yielding Phosphoinositol-3,4,5 tris Phosphate (PIP3).
Phosphatidyl Inositol Dependent Protein Kinase 1 - PDK 1 - and Protein Kinase B/AKT - PKB/AKT - and Atypical Protein Kinase C - aPKC - each bind to a different molecule of PIP3 by their "Plekstrin Homology" domains, bringing them into close proximity to one another and activating the PDK1 protein kinase activity. PDK 1 phosphorylates both PKB/AKT and aPKc, which activates their respective protein kinase activities.
Activated PKB/AKT and aPKc dissociated from PIP3 to find and phosphorylate their target proteins in the cytoplasm. The target proteins mediate the effects of insulin on glucose metabolism, including GLUT4 mobilization to the plasma membrane, activation of protein phosphatases that reverse the effects of glucagon, e.g., the synthesis of glucokinase, phosphoenolpyruvate carboxykinase, fructose-1,6-bisphosphatase and glucose-6 phosphatase. Phosphorylation by PK B/Akt and/or aPK C also mediates the affects of insulin on several other cellular processes, including protein synthesis, cell growth, cell cycle entry and cell survival.
The glucagon receptor is a member of the family of heptahelical G-protein-coupled receptors, which have seven α-helical membrane spanning domains. Although hundreds of hormones and neurotransmitters function through heptahelical receptors, their extracellular binding domains are specific for only one polypeptide hormone, catecholamine, or neurotransmitter. Heptahelical receptors have no intrinsic protein kinase activity but initiate signal transduction by their intracellular interaction with trimeric G-proteins (guanine nucleotide-binding proteins) composed of α, β, and γ subunits. Different types of heptahelical receptors bind different types of G-proteins, which exert different effects on their target proteins.
| G-protein Type | Known Functions |
| G α s | stimiulates the production of cAMP from ATP. This is accomplished by direct stimulation of the membrane-associated enzyme adenylate cyclase. cAMP acts as a second messenger that interacts with and activates protein kinase A (PKA), also known as cyclic AMP-dependent Protein Kinase. PKA can then phosphorylate a myriad of downstream targets. |
| G α i | inhibits the production of cAMP from ATP |
| G α q/11 | stimulates membrane-bound phospholipase C β, which then cleaves PIP2 (a minor membrane phosphoinositol) into two second messengers: IP3 and diacylglycerol (DAG) |
| G α 12/13 | are involved in Rho family GTPases signaling 9through the RhoGEF superfamily) and control cell sytoskeleton remodeling thus regulating cell migration |
| G β γ | G β γ subunits of G-proteins sometimes also have active functions, e.g., coupling to L-type calciium channels |
The un-liganded receptor has a conformation that does not activate the trimeric Gs protein, which binds GDP.
Gucagon binding induces a conformational change in the receptor allowing it to activate the trimeric Gs protein. This activation induces the exchange of GDP for GTP and the release of the α subunit from the β/γ subunits.
The α subiunit is free to move in the plasma membrane and when it encounters a molecule of adenylyl cyclase it binds to it and activates it to convert ATP to cAMP with the release of inorganic pyrophosphate (PPi).
An endogenous, slow acting GTPase in the α subunit hydrolyses the GTP to GDP, thereby inactivating the α subunit, which dissociates from the adenylyl cyclase.
The inactive α subunit bound to GDP reassociates with the β/γ subunits and the unbound adenylyl cyclase returns to its inactive state.
The extracellular domain of the β2 Adrenergic Receptor receptor binds epinephrine/adrenaline (not glucagon) and transmits a signal through the plasma membrane to the same trimeric G protein as does the glucagon receptor; the same intracellular signaling pathway ensues, causing an increase in intracellular cAMP.
The extracellular domain of the GLP-1 receptor binds the GLP-1 peptide (not either glucagon nor epinephrine) and transmits a signal through the plasma membrane to the same trimeric G protein as does the glucagon receptor and the β2 Adrenergic Receptor; the same intracellular signaling pathway ensues, causing an increase in intracellular cAMP.
In the absence or low concentration of cAMP, the PKA catalytic subuints are bound by the PKA regulatory subunit, thereby sequestering them.
In response to glucagon signaling (also epinephrine signaling or GPL-1 signaling), the concentration of cAMP increases.
Binding of cAMP to the PKA regulatory subunit induces a conformational alteration of the regulatory subunit such that it cannot bind to the PKA catalytic subunits, which it releases. The active catalytic subunits phosphorylate their target proteins, either activating or inactivating them.
As signaling at the cell surface receptor declines, the intraellular concentration of cAMP also declines, reversing the conformational change of the PKA regulatory subunit and causing the catalytic subunits to rejoin the regulatory subunit.
The protein kinase A catalytic subunits are sequestered in the absence of cAMP.
The increased intracellular concentration of cAMP activates cyclic AMP-dependent protein kinase, also called protein kinase A (PKA), a serine/threonine protein kinase, by binding to its repressive regulatory subunits, which then undergo a conformation change and release the active PKA catalytic subunits. PKA phosphorylates a large number of metabolic enzymes, thereby providing a rapid response to hormones such as glucagon and epinephrine, which, when it binds to the β2-adronergic receptor, induces the same intracellular trimeric G-protein/adenylyl cyclase pathway as does glucagon to increase cAMP levels.
The signaling process is reversed when the secretion of glucagon, which has a half-life in the blood of approximately 4 minutes, declines. The Gα subunit of the Gs trimeric G-protein becomes inactive as a result of its GTPase activity, and remains inactive in the absence of activation by glucagon. cAMP is hydrolyzed to AMP by cAMP phosphodiesterase, thereby reducing the levls of cAMP and causing the PKA catalytic subunits to rejoin the repressive regulatory subunits. The concentration of cAMP and other second messengers is balanced by the activities of those processes that synthesize them and those processes that destroy them. In the presence of hormone, cAMP synthesis exceeds cAMP destruction and cAMP concentration increases. In the absence of hormone cAMP destruction exceeds cAMP synthesis and cAMP concentration decreases.
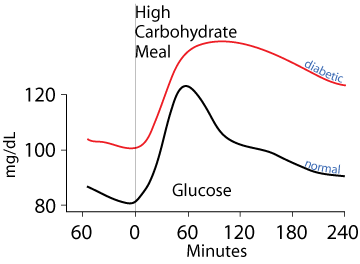
Untreated diabetics fail to clear glucose from the blood normally either due to a lack of insulin (Type 1 diabetes) or failure of tissues that normally clear glucose from the blood to respond to insulin signaling (Type 2 diabetes).
Type 1 diabetes results from the autoimmune destruction of the pancreatic β cells, causing the loss of insulin production. Type 2 diabetes is the resistance of target cells to signaling by insulin. Both Type 1 and Type 2 diabetes result in failure to normally clear dietary glucose from the blood and to abnormally synthesize glucose from non-carbohydrate carbon skeletons and export it into the blood, even when blood [glucose] is abnormally high. The classic symptoms are frequent urination and increased thirst due to the osmotic activity of excess glucose in the blood and urine, which draws water from tissues into the blood and urine, increased hunger due to failure to normally process food molecules, and weight loss due to the failure to transition normally between the catabolic (fasted) and anabolic (fed) state, remaining constantly in the catabolic state. Additional symptoms may include blurry vision and poor wound healing due to the damage of functional proteins by their abnormal non-enzymatic glycosylation resulting from the chemical reactivity of glucose.
Untreated Type 2 diabetes ultimately progresses to Type 1 diabetes because the failure to clear glucose from the blood causes the β cells to continually attempt to synthesize and secrete insulin, resulting in endoplasmic reticulum stress (ER stress) from which the β cells eventually die.